The Third-Party (3P) Review Program is authorized under Section 523 of the Federal Food, Drug, and Cosmetic Act. It allows FDA-accredited third-party organizations (3P510k) to review 510(k) submissions for certain low-to-moderate risk medical devices.
Under the program, a medical device manufacturer forwards its 510(k) submission for a qualifying device to a 3PRO instead of to the FDA. The 3PRO reviews the submission utilizing the same standards as the FDA and then prepares a review of the submission and a recommendation, which are then forwarded to the FDA for a final decision.
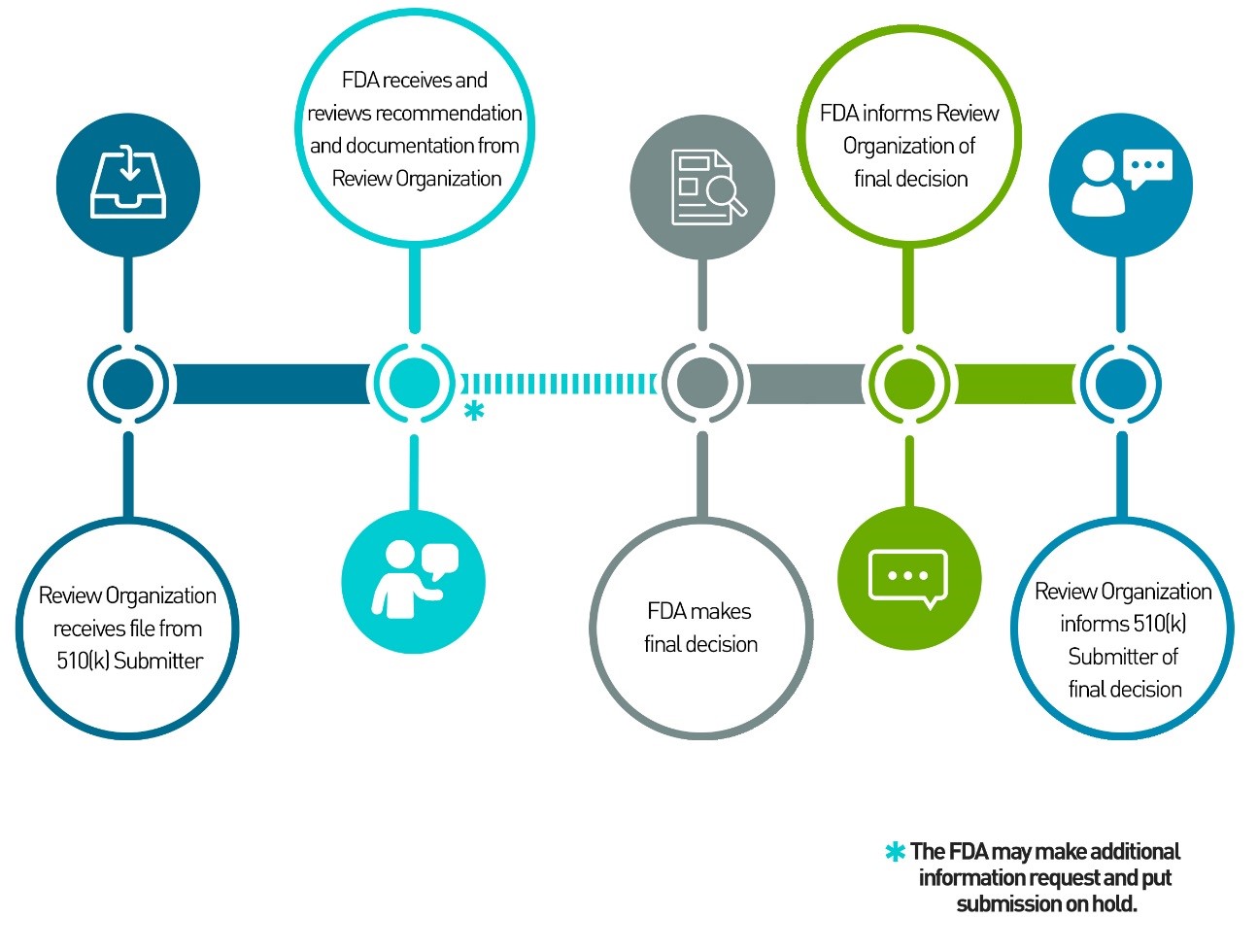
Previously, the FDA re-reviewed all 510(k) submissions sent in by 3PROs before considering their review and recommendation. The agency has revised that practice and now no longer routinely re-reviews the 3P 510(k) submissions. Instead, it relies on the review and recommendation of the 3PRO before deciding whether a product is Substantially Equivalent (SE) or Not Substantially Equivalent (NSE).
The modification to the FDA’s review policy now enables the agency to make a final determination within 30 days of receiving the 3PRO’s review and recommendation. That compares to the 90 days the FDA is permitted when a 510(k) submission is sent directly to the agency for review.
The FDA has also been given legislative authority to tailor the list of devices eligible for review by 3PROs. As a result, some of the more complex devices that have historically required additional levels of FDA review have been removed from the program while other less-complex devices have been added.
The FDA is also providing 3PRO reviewers with more of the tools they need to perform FDA-equivalent reviews. This includes training programs that are comparable to those available to agency reviewers, memo templates tailored to specific device types, and timely updates outlining adjustments the FDA has made to its review practices due to changes in medical device technology.